Helps medical researchers to determine the somatic status of each variant, to identify and filters and detects germline variants for parallel sequencing. #Identify snp #Sample seqeuencer #Snp identifier #Identifier #SN: #Sequence
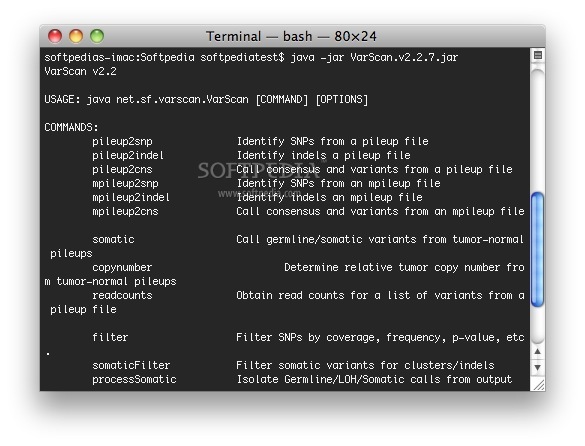
VarScan is a free and open source, platform-independent, technology-independent software tool for identifying SNPs and deals in massively parallel sequencing of individual and pooled samples.
Given data for a single sample, VarScan identifies and filters germline variants based on read counts, base quality, and allele frequencyh.
Given data for a tumor-normal pair, VarScan also determines the somatic status of each variant (Germline, Somatic, or LOH) by comparing read counts between samples.
Variant detection in massively parallel sequencing. For one sample, calls SNPs, indels, and consensus genotypes. For tumor-normal pairs, further classifies each variant as Germline, Somatic, or LOH, and also detects somatic copy number changes.
System requirements
VarScan 2.3.8
add to watchlist add to download basket send us an update REPORT- runs on:
- Mac OS X (PPC & Intel)
- file size:
- 111 KB
1 screenshot:
- main category:
- Math/Scientific
- developer:
- visit homepage
calibre
Effortlessly keep your e-book library thoroughly organized with the help of the numerous features offered by this efficient and capable manager

4k Video Downloader
Export your favorite YouTube videos and playlists with this intuitive, lightweight program, built to facilitate downloading clips from the popular website

Bitdefender Antivirus Free
Feather-light and free antivirus solution from renowned developer that keeps the PC protected at all times from malware without requiring user configuration

Microsoft Teams
Effortlessly chat, collaborate on projects, and transfer files within a business-like environment by employing this Microsoft-vetted application

Context Menu Manager
Customize Windows’ original right-click context menu using this free, portable and open-source utility meant to enhance your workflow

Windows Sandbox Launcher
Set up the Windows Sandbox parameters to your specific requirements, with this dedicated launcher that features advanced parametrization

7-Zip
An intuitive application with a very good compression ratio that can help you not only create and extract archives, but also test them for errors

ShareX
Capture your screen, create GIFs, and record videos through this versatile solution that includes various other amenities: an OCR scanner, image uploader, URL shortener, and much more

Zoom Client
The official desktop client for Zoom, the popular video conferencing and collaboration tool used by millions of people worldwide

IrfanView
With support for a long list of plugins, this minimalistic utility helps you view images, as well as edit and convert them using a built-in batch mode

% discount
ShareX
- ShareX
- Zoom Client
- IrfanView
- calibre
- 4k Video Downloader
- Bitdefender Antivirus Free
- Microsoft Teams
- Context Menu Manager
- Windows Sandbox Launcher
- 7-Zip
essentials
User Comments
This enables Disqus, Inc. to process some of your data. Disqus privacy policy